The US FDA issued an emergency use authorization (EUA) for Regeneron’s casirivimab and imdevimab to be administered together for the treatment of mild to moderate COVID-19 in adults and pediatric patients (12 years of age or older weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progressing to severe COVID-19. This includes those who are 65 years of age or older or who have certain chronic medical conditions.
In a clinical trial of patients with COVID-19, casirivimab and imdevimab, administered together, were shown to reduce COVID-19-related hospitalization or emergency room visits in patients at high risk for disease progression within 28 days after treatment when compared to placebo. The safety and effectiveness of this investigational therapy for use in the treatment of COVID-19 continues to be evaluated.
Casirivimab and imdevimab must be administered together by intravenous (IV) infusion.
Casirivimab and imdevimab are not authorized for patients who are hospitalized due to COVID-19 or require oxygen therapy due to COVID-19. A benefit of casirivimab and imdevimab treatment has not been shown in patients hospitalized due to COVID-19. Monoclonal antibodies, such as casirivimab and imdevimab, may be associated with worse clinical outcomes when administered to hospitalized patients with COVID-19 requiring high flow oxygen or mechanical ventilation.
“The FDA remains committed to advancing the nation’s public health during this unprecedented pandemic. Authorizing these monoclonal antibody therapies may help outpatients avoid hospitalization and alleviate the burden on our health care system,” said FDA Commissioner Stephen M. Hahn, MD. “As part of our Coronavirus Treatment Acceleration Program, the FDA uses every possible pathway to make new treatments available to patients as quickly as possible while continuing to study the safety and effectiveness of these treatments.”
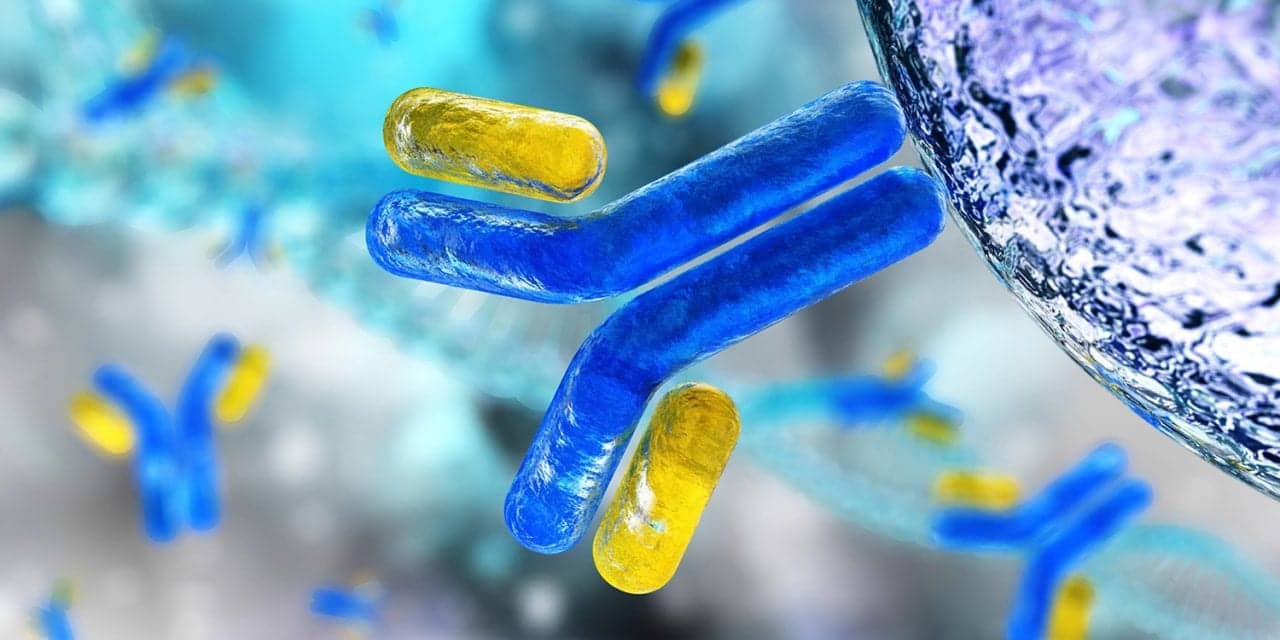
Monoclonal antibodies are laboratory-made proteins that mimic the immune system’s ability to fight off harmful pathogens such as viruses. Casirivimab and imdevimab are monoclonal antibodies that are specifically directed against the spike protein of SARS-CoV-2, designed to block the virus’ attachment and entry into human cells.
“The emergency authorization of these monoclonal antibodies administered together offers health care providers another tool in combating the pandemic,” said Patrizia Cavazzoni, MD, acting director of the FDA’s Center for Drug Evaluation and Research. “We will continue to facilitate the development, evaluation and availability of COVID-19 therapies.”
The issuance of an EUA is different than an FDA approval. In determining whether to issue an EUA, the FDA evaluates the totality of available scientific evidence and carefully balances any known or potential risks with any known or potential benefits of the product for use during an emergency. Based on the FDA’s review of the totality of the scientific evidence available, the agency has determined that it is reasonable to believe that casirivimab and imdevimab administered together may be effective in treating patients with mild or moderate COVID-19. When used to treat COVID-19 for the authorized population, the known and potential benefits of these antibodies outweigh the known and potential risks. There are no adequate, approved and available alternative treatments to casirivimab and imdevimab administered together for the authorized population.
The data supporting this EUA for casirivimab and imdevimab are based on a randomized, double-blind, placebo-controlled clinical trial in 799 non-hospitalized adults with mild to moderate COVID-19 symptoms. Of these patients, 266 received a single intravenous infusion of 2,400 milligrams casirivimab and imdevimab (1,200 mg of each), 267 received 8,000 mg casirivimab and imdevimab (4,000 mg of each), and 266 received a placebo, within three days of obtaining a positive SARS-CoV-2 viral test.
The prespecified primary endpoint for the trial was time-weighted average change in viral load from baseline. Viral load reduction in patients treated with casirivimab and imdevimab was larger than in patients treated with placebo at day seven. However, the most important evidence that casirivimab and imdevimab administered together may be effective came from the predefined secondary endpoint of medically attended visits related to COVID-19, particularly hospitalizations and emergency room visits within 28 days after treatment. For patients at high risk for disease progression, hospitalizations and emergency room visits occurred in 3% of casirivimab and imdevimab-treated patients on average compared to 9% in placebo-treated patients. The effects on viral load, reduction in hospitalizations and ER visits were similar in patients receiving either of the two casirivimab and imdevimab doses.
Under the EUA, fact sheets that provide important information about using casirivimab and imdevimab administered together in treating COVID-19 as authorized must be made available to healthcare providers and to patients and caregivers. These fact sheets include dosing instructions, potential side effects and drug interactions. Possible side effects of casirivimab and imdevimab include: anaphylaxis and infusion-related reactions, fever, chills, hives, itching and flushing.