 Using genome sequencing, National Institutes of Health (NIH) scientists and their colleagues have tracked the evolution of the antibiotic-resistant bacterium Klebsiella pneumoniae sequence type 258 (ST258), an important agent of hospital-acquired infections.
Using genome sequencing, National Institutes of Health (NIH) scientists and their colleagues have tracked the evolution of the antibiotic-resistant bacterium Klebsiella pneumoniae sequence type 258 (ST258), an important agent of hospital-acquired infections.
While researchers had previously thought that ST258 K. pneumoniae strains spread from a single ancestor, the NIH team showed that the strains arose from at least two different lineages. The investigators also found that the key difference between the two groups lies in the genes involved in production of the bacterium’s outer coat, the primary region that interacts with the human immune system.
Their results, which appear online in Proceedings of the National Academy of Sciences, promise to help guide the development of new strategies to diagnose, prevent, and treat this emerging public health threat.
ST258 K. pneumoniae is the predominant cause of human infections among bacteria classified as carbapenem-resistant Enterobacteriaceae (CRE), which kill approximately 600 people annually in the United States and sicken thousands more. Most CRE infections occur in hospitals and long-term care facilities among patients who are already weakened by unrelated disease or have undergone certain medical procedures.
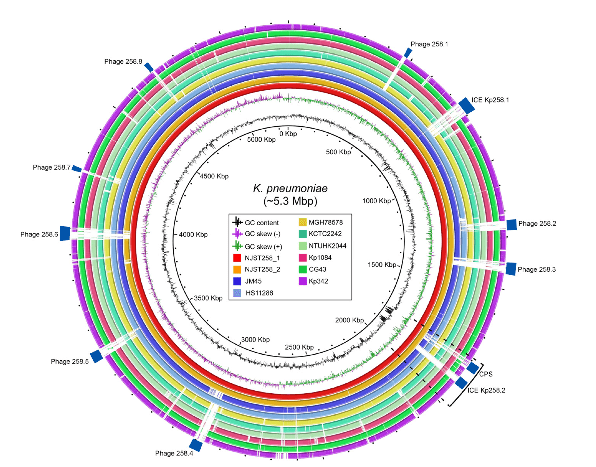
In the new study, scientists from the NIH’s National Institute of Allergy and Infectious Diseases (NIAID) and their colleagues sequenced the complete genomes of ST258 K. pneumoniae strains collected from two patients in New Jersey hospitals. By comparing these reference genomes with gene sequences from an additional 83 clinical ST258 K. pneumoniae isolates, the scientists found that the strains divided broadly into two distinct groups, each with its own evolutionary history.
Further analysis revealed that most differences between the two groups occur in a single “hotspot” of the genome containing genes that produce parts of the bacterium’s outer shell. The investigators plan to further study how these genetic differences may affect the bacterium’s ability to evade the human immune system.
The findings from this study highlight the wealth of information that can be gained from genome sequencing. They also demonstrate the importance of sequencing to the surveillance and accurate tracking of bacterial spread. Study collaborators included NIAID-funded scientists from Public Health Research Institute and New Jersey Medical School-Rutgers University, as well as researchers from Case Western Reserve University, the Houston Methodist Research Institute and Hospital System and NIAID’s Rocky Mountain Laboratories, where the comparative genome sequencing took place.