Acid-base disorders can be an indication of serious disease, and it is important that clinicians identify the cause before embarking on a treatment plan.
By Rick Carter, PhD, MBA; Brian Tiep, MD; and Doug Boatright, PhD
Numerous physiological and pathological processes alter blood gases and acid-base status. It is critical that oxygen delivery be maintained at adequate physiologic levels based on organ and cellular demand. Any fluctuation from the normal physiological range may compromise the systems and eventually lead to cell destruction and death. Thus, when interpreting arterial blood gas data, it is important that the clinician have available sufficient data to construct a clinical picture of oxygen delivery and metabolic status.1 The data must be accurate and free from artifact and technical error.
Blood-gas data will aid clinical decision-making and influence therapeutic modalities. The basic underlying goal is to maximize oxygen delivery to the tissues and correct the underlying pathophysiologic processes.
Oxygen Delivery
Hypoxemia may be the result of inadequate alveolar ventilation VA/Q mismatch, impaired diffusing capacity, or arterial to venous shunt.1 Hypoxemia often leads to tissue hypoxia, and correction of hypoxemia may help to reverse tissue hypoxia.2 There are also several other causes of tissue hypoxia, many of which we lack specific tools to measure. Ideally, it would be desirable to measure the oxygen status of each organ. Short of that ability, PaO2, mixed venous PO2 (PvO2), hemoglobin (Hb), and cardiac output (Q) are measured in the blood gas laboratory. These variables provide important insights regarding oxygen delivery (DO2).
Acid-Base Disorders
Acid-base equilibrium may be disrupted in a wide variety of chronic and critical illnesses and is among the commonly encountered disorders in the intensive care unit (ICU).2 Management of diverse disorders ranges from diabetic ketoacidosis to tissue hypoperfusion with lactic acidosis resulting from hemorrhagic or septic shock, with each sharing common therapies for correction of acid-base balance.
Not only do acid-base disorders reflect the seriousness of the underlying pathophysiologic process(s), these disorders have their own associated morbidity and mortality.2 Therefore, it is imperative that clinicians search for and identify the cause for acid-base shifts rather than attempt to normalize pH. Mild shifts in acid-base balance may not require a prescription while more significant shifts may exacerbate the course of the disease process. In order for a treatment plan to be established, the underlying cause should be identified and the many tools available to dissect the true nature of the acid-base disturbance should be emphasized.
Physiologic Effects
Physiologic systems rely on a balance among the different ions that are completely dissociated at physiologic pH. The strong cations (Na+, K+, Ca+, and Mg+), as well as the strong anions (Cl–, lactate, and sulfates [most notable during renal failure]), help establish an equilibrium and thus create a strong ion difference (SID[+40]).3 According to physical chemistry principles, the difference must be counterbalanced by an equal and opposing charge termed the effective strong ion difference (SIDe [-40]), and this negative charge comes from dissociated moieties of plasma proteins albumin (78%) and phosphate (20%).4 When the SID and SIDe are equal, the plasma pH is at 7.4 at a Pco2 of 40 mm Hg.
Acidemia can lead to decreased cardiac contractility that is directly proportional to the decrease in pH experienced. While both metabolic and respiratory acidemia can depress the cardiac myocyte contractile function, respiratory acidemia is known to occur more promptly, and this is attributed to CO2‘s rapid entry into the cardiac myocyte.5 While animal studies have demonstrated a lowering of the threshold for ventricular fibrillation with acidemia, clinically no significant increase in arrhythmias is seen, and there appears to be no detrimental effect in successful defibrillation. Acidemia stimulates the sympathetic-adrenal axis, and, when severe, this effect is countered by a depressed responsiveness of adrenergic receptors to circulating catecholamines. Moreover, because the acidemia resulting from hypercapnia is globally distributed throughout the body, other symptoms may be seen, including depression of diaphragmatic contractility or decreased endurance time.
Cerebral blood flow is compromised in acute respiratory acidemia, and when Paco2 rises to 60 mm Hg or more, confusion and headache ensue.6 In the event that 70 mm Hg Paco2 thresholds are encountered, a loss of consciousness and seizures can arise. Chronic elevations in CO2 are usually well tolerated even to Paco2 values up to 150 mm Hg in the presence of full metabolic (HCO3–) compensation. Thus, symptomatology differs greatly between acute and chronic CO2 retention, and the therapeutic approach followed must take into account the underlying pathophysiology and the rapidity of presentation.
The effects of acidemia on electrolyte levels are complex. Acute infusion of HCl causes an increase in serum potassium, yet administration of organic acids (lactic and ketoacids) does not elevate serum potassium and may even lower their values.7 Hyperkalemia observed in both lactic acidosis and ketoacidosis is not due to pH shifts but other compensatory mechanisms. Acute respiratory acidemia causes a slight or no change in serum potassium while both respiratory and metabolic acidosis cause increased extracellular phosphate concentrations. In addition to hyperkalemia, lactic acidosis and ketoacidosis are also associated with hyperphosphatemia.
Alkalemia has been shown to increase cardiac myocyte contractility up to a pH of 7.7 with little if any effect on ventricular fibrillation threshold.8 Systemic vascular resistance is lowered during hyperventilation. Alkalemia is associated with coronary artery spasm with ECG evidence of ischemia, and hyperventilation with alkalemia has been used to provoke coronary vasospasm.9
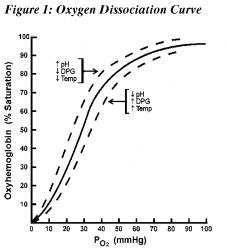
Acute alkalemia is associated with decreased cerebral blood flow, leading to symptoms of confusion, myoclonus, asterixis, loss of consciousness, and seizures.10 Acute hypocapnia causes slight reductions in serum sodium, potassium, and phosphorus. Alkalemia shifts the oxygen disassociation curve to the left, increasing hemoglobin’s affinity for oxygen (Figure 1).11 Compensatory changes in the concentration of 2,3 DPG in red blood cells and morphological changes counter left shift O2 loading. 2,3 DPG is a by-product of red blood cell anaerobic glycolysis, which can combine with the hemoglobin and reduce hemoglobin’s affinity for oxygen (right shift).11 Thus, the overall effects of alkalemia on oxygen delivery are often minimal; however, they may be important for patients with tissue hypoxia.
Working with Acid-Base Decision Pathways
Paco2 represents the patient’s ventilatory status and is the respiratory component of acid-base balance. CO2 is a volatile acid and represents a large fraction of the daily acid production by the body. Further, CO2 is a by-product of glycolysis and is converted to carbonic acid in red blood cells by carbonic anhydrase (CA).1

HCO3– exits the red blood cell in large quantities because of hemoglobin’s ability to buffer protons produced by the reaction above. A metabolic chloride-HCO3– exchange mechanism is responsible for maintaining this HCO3– gradient, as well as for producing the chloride shift (Figure 2).

These chemical reactions promote transport of CO2 with about 66% of the metabolically produced CO2 transported in plasma as HCO3–, 8% as dissolved CO2, and the remaining 25% transported by Hb or carbamino groups.12 These mechanisms allow for the transport of large amounts of CO2 in blood without creating a large CO2 gradient and without significantly lowering the blood pH.
HCO3– represents the metabolic component for acid-base equilibrium and is continuously adjusted by the kidneys.13 The kidneys have the option to excrete less or more HCO3–, lowering the pH and increasing acidity or retaining HCO3–, thereby raising the pH with the blood becoming more alkaline. It is important to remember that this process takes several hours to days.
Acid-base disorders affect changes in arterial Pco2, serum HCO3, and pH. The normal pH range for the human body is 7.35 to 7.45; beyond either limit being categorized as acidemia (low pH) or alkalemia (high pH).14 Acidosis is the physiologic process that causes acid accumulation or alkali loss whereas alkalosis is the accumulation of alkali or acid loss. The actual changes in pH depend on the acute process, as well as physiologic compensatory processes.
The primary acid-base disorders can be classified as metabolic or respiratory, based on clinical presentation and whether the prevailing pH change is the result of HCO3– or Pco2. Metabolic acidosis is a serum HCO3– <24 mEq/L and is caused by increased acid production, acid ingestion, decreased renal acid excretion, and GI or renal HCO3– loss. Acidosis from decreased renal excretion is generally slow to develop, yet acidosis from lactate acidosis or ketoacidosis can exceed renal excretion and cause a rapidly developing, severe acidosis.1 The etiologies of metabolic acidosis can be discerned by those that cause an increase in the anion gap, which is the difference between measured cations and measured anions as defined as:
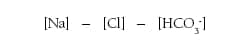
The normal range for the anion gap in healthy adults ranges from 8 to 12 mEq/L, and abnormal values are defined as two standard deviations above or below the average level, thus the upper bound is 11 to 12 mEq/L.15 Different measurement techniques alter the normal value ranges; therefore, it is important for the clinician to understand the measurement technique. Most blood gas laboratories use the calculated anion gap with or without K+ present, eg, ([Na+]+[K+]) – ([Cl–]+[HCO3–]) or ([Na+]) – ([Cl–]+[HCO3–]). Yet, there are more precise calculations that can be applied that consider other ions such as Mg2+ and Ca2+, while applying a correction for the concentration of albumin and phosphate, eg:
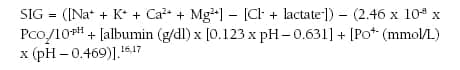
A normal anion gap occurs when Cl– replaces HCO3– lost in buffering H. An increased anion gap acidosis occurs when the anion replacing the HCO3– is not one that is routinely measured (albumin, phosphate, sulfates, lactate, and others).18-21 Anions always equal cations, but if the anion is not Cl–, then the anion gap calculated from routine chemistries will increase. Yet, an increased anion gap does not always signify a metabolic acidosis. The anion gap is increased in alkalemia, resulting from a net anionic charge on the plasma proteins. Dehydration will increase the plasma protein net anionic charge, and, if the anion gap is greater than 20 mEq/L, a metabolic acidosis should be considered.
The anion gap is usually classified as low, within normal limits, or high.22,23 From a blood collection and assay perspective, it is important that the blood sample be assayed as soon as possible, because ongoing leukocyte metabolism may result in an increase in HCO3– with a mild reduction in the anion gap. To counter this effect, icing of the blood sample is recommended. Changes in the anion gap can result from renal function changes (even mild such as that induced by diarrhea)24 and alter the expected change from a particular pathophysiologic process.
A high anion gap results from a loss in HCO3– without a concurrent increase in Cl–. Elevated levels of lactate, beta-hydroxybutyrate and acetoacetate, PO4–, and SO4– maintain electroneutrality, because these anions are not part of the anion-gap calculation and high anion-gap results. Therefore, a search for pathophysiologic processes that produce an excess of these anions should be sought. A high anion gap is noted for: lactic acidosis, ketoacidosis (diabetic ketoacidosis, alcohol abuse), toxins (ethylene glycol, lactic acid, methanol, propylene glycol—common in pharmaceutical injections such as diazepam or lorazepam, phenformin, aspirin, cyanide, iron, and isoniazid). A normal gap may result from HCO3– loss that is replaced by Cl–. This can occur with GI loss (diarrhea); renal loss (proximal renal tubular acidosis, RTA); renal dysfunction (renal failure, hypoaldosteronism, or distal renal tubular acidosis); through ingestion of ammonium chloride, acetazolamide, or hyperalimentation fluids (TPN); through some cases of ketoacidosis, particularly during rehydration with Na+ IV solutions; or alcohol (high anion gap or mixed in others due to concurrent metabolic alkalosis or through mineralocorticoid deficiency).25,26 A low anion gap is largely the result of hypoalbuminemia (albumin is negatively charged and its loss from serum promotes retention of negatively charged ions such as Cl– and HCO3–.27 For hypoalbuminemia, the anion gap is reduced from between 2.5 and 3.0 mmol/L per g/dL decrease in albumin, which can result from hemorrhage, nephritic syndrome, intestinal obstruction, or liver cirrhosis.19 Additionally, multiple myeloma with increased IgG (paraproteinemia) may elicit a low anion gap.10
Patients admitted to the ICU commonly experience lactate acidosis, which has significant physiologic implications and serves as a marker for serious underlying pathophysiologic processes. Lactic acidosis is commonly defined as a lactate level greater than 5 mmol/L, with an arterial pH less than 7.35.1 Increased lactate levels in cardiogenic shock are correlated with mortality, yet this relationship decreases with other forms of shock. The lactate trend for a patient can be helpful in gauging the effect of therapy and establishing a prognosis.
Metabolic alkalosis, on the other hand, is when the HCO3– >24 mEq/L and results from acid loss or HCO3– retention.1 Respiratory acidosis is when the Pco2 >45+ mm Hg (hypercapnia) and results from hypoventilation, while respiratory alkalosis is a Pco2 <35- mm Hg and is the result of hyperventilation. Whenever acid-base disorders are present, compensatory mechanisms initiate a change in serum pH (Table 1). Physiologic compensation cannot return pH to a completely normal value and does not overshoot the anticipated target.

Summary
Understanding the principles and complexities of acid-base disorders is essential for the clinician. High-quality care begins with a thorough understanding of the physiology and pathophysiology dictating movement with the specific index. This is coupled to a thorough understanding of collection pitfalls and errors with respect to ABG analysis and quality control. To differentiate among the numerous pathophysiologies that may present, additional laboratory data or calculations may be required. This can include the anion gap and base excess calculations, the various forms of hemoglobin, and other clinical chemistries. Each index has its advantages and limitations. Thus, it is imperative that laboratory personnel understand the pros and cons associated with each index and be available to address the nuances of each.
RT
Rick Carter, PhD, MBA, is professor of health and exercise sciences at Lamar University, Beaumont, Tex. Prior to this appointment, he spent 23 years at The University of Texas Health Science Center in Tyler, where he was professor of medicine and physiology and directed several clinical units.
Brian Tiep, MD, is the program designer and medical director of the Respiratory Disease Management Institute in Pomona and Irwindale, Calif; associate professor at Western University of Health Science in Pomona; and director of pulmonary rehabilitation at City of Hope Cancer Center, Duarte, Calif.
Doug Boatright, PhD, is professor of health and exercise sciences at Lamar University.
References
- Carter R. Oxygen and acid-base status: measurement, interpretation, and rationale for oxygen therapy. In: Tiep BL, ed. Portable Oxygen Therapy: Including Oxygen Conserving Methodology. Mount Kisco, NY: Futura Publishing Co; 1991:125-59.
- Fishman AF. Pulmonary Diseases and Disorders. New York: McGraw Hill; 1991.
- Boniatti MM, Cardoso PR, Castilho RK, et al. Acid-base disorders evaluation in critically ill patients: we can improve our diagnostic ability. Intensive Care Med. 2009;35:1377-82.
- Barach AL. Home rehabilitation for pulmonary emphysema: device for delivering oxygen during inspiratory cycle. N Y State J Med. 1970;70:71-82.
- Colwell HH, Mathias SD, Pasta DJ, et al. Development of a health-related quality-of-life questionnaire for individuals with gastroesophageal reflux disease: a validation study. Dig Dis Sci. 1999;44:1376-83.
- Lambertson CJ. In: Mountcastle VB, ed. Medical Physiology. 13th ed. St Louis: Mosby Publishing; 1974:1447-1521.
- Gossett KA, French DD, Cleghorn B, Church GE. Effect of acute acidemia on blood biochemical variables in healthy ponies. Am J Vet Res. 1990;5:1375-9.
- Turnbull AD, Dobell AR. The effect of pH change on the ventricular fibrillation threshold. Surgery. 1966;60:1040-3.
- Dong E Jr, Stinson EB, Shumway NE. The ventricular fibrillation threshold in respiratory acidosis and alkalosis. Surgery. 1967;6:602-7.
- Herd AA. An approach to complex acid-base problems: keeping it simple. Can Fam Physician. 2005;51:226-32.
- Lambertson CJ. Transport of oxygen and carbon dioxide by the blood. In: Mountcastle V, ed. Medical Physiology. St Louis: Mosby; 1974:1399-1422.
- Adams AP, Hahn CEW. Principles and Practice of Blood-gas Analysis. London: Franklin Scientific Projects Ltd; 1979.
- Carter R. The physiologic principles of oxygen delivery. In: Tiep BL, ed. Portable Oxygen Therapy: Including Oxygen Conserving Methodology. Mount Kisco, NY: Futura Publishing Co; 1991:81-124.
- de Morais HA, Bach JF, DiBartola SP. Metabolic acid-base disorders in the critical care unit. Vet Clin North Am Small Anim Pract. 2008;38:559-74, x-xi.
- Elisaf MS, Siamopoulos KC. Serum anion gap: reevaluation of normal values. Br J Clin Pract. 1996;50-411.
- Kellum JA, Kramer DJ, Pinksy MR. Strong anion gap: a methodology for exploring unexplained anions. J Crit Care. 1995;10:51-5.
- Kellum JA, Bellomo R, Kramer DJ, Pinsky MR. Hepatic anion flux during acute endotoxemia. J Appl Physiol. 1995;78:2212-17.
- Lolekha PH, Lolekha S. Value of the anion gap in clinical diagnosis and laboratory evaluation. Clin Chem. 1983;29:279-83.
- Feldman M, Soni N, Dickson B. Influence of hypoalbuminemia or hyperalbuminemia on the serum anion gap. J Lab Clin Med. 2005;146:317-20.
- Kraut JA, Madias NE. Serum anion gap: its uses and limitations in clinical medicine. Clin J Am Soc Nephrol. 2007;2:162-74.
- Bouchard J, Mehta RL. Acid-base disturbances in the intensive care unit: current issues and the use of continuous renal replacement therapy as a customized treatment tool. Int J Artif Organs. 2008;31:6-14.
- Chawla LS, Jagasia D, Abell LM, et al. Anion gap, anion gap corrected for albumin, and base deficit fail to accurately diagnose clinically significant hyperlactatemia in critically ill patients. J Intensive Care Med. 2008;23:122-7.
- Reddy P, Mooradian AD. Clinical utility of anion gap in deciphering acid-base disorders. Int J Clin Pract. 2009;63:1516-25.
- Edwards SL. Pathophysiology of acid base balance: the theory practice relationship. Intensive Crit Care Nursing. 2008;24:28-38.
- Zietse R, Zoutendijk R, Hoorn EJ. Fluid, electrolyte and acid-base disorders associated with antibiotic therapy. Nat Rev Nephrol. 2009;5:193-202.
- Alcazar AR. [Electrolyte and acid-base balance disorders in advanced chronic kidney disease.] Nefrologia. 2008;28(Suppl 3):87-93.
- Fidkowski C, Helstrom J. Diagnosing metabolic acidosis in the critically ill: bridging the anion gap, Stewart, and base excess methods. Can J Anaesth. 2009;56:247-56.