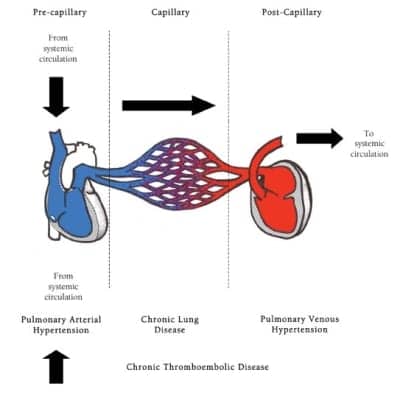

Figure. Pulmonary Circulation.
Pulmonary hypertension (PH) is the end result of a plethora of diseases that cause increased resistance in the pulmonary vasculature. The elevated pressures typically are found to be precapillary (known as pulmonary arterial hypertension, or PAH), postcapillary (pulmonary venous hypertension as in left heart failure), or capillary (as in chronic lung diseases), or from chronic thromboembolic diseases (see Figure).
Patients with PH are clinically categorized into four classes based on the New York Heart Association classification categories for heart failure.1 They range from asymptomatic (Class I) to extreme fatigue and breathlessness at rest (Class IV). Unfortunately, many PH patients will be a Class III or IV when they are diagnosed due to the disease’s insidious course and nonspecific signs and symptoms.
Historically, PH has been defined by a right-sided cardiac catheterization, which remains the gold standard for diagnosis.2 A mean pulmonary artery pressure of greater than 25 mm Hg at rest or greater than 30 mm Hg during exercise with a normal pulmonary capillary wedge pressure (PCWP) and an elevated pulmonary vascular resistance greater than 3 Wood units characterize PAH. With pulmonary venous hypertension, the PCWP is typically elevated to >15 mm Hg.
Symptoms
Symptoms associated with PH include shortness of breath, first with exertion and subsequently at rest; peripheral edema; decreased exercise tolerance; dizziness; and fatigue. Echocardiographic signs include tricuspid regurgitation with elevated right ventricular systolic pressure and an enlarged right ventricle with decreased contractility.3 Classically, patients with pulmonary vascular disease on pulmonary function testing will show a decrease in their Dlco with otherwise normal spirometry and lung volumes.

Table 1. Diagnostic tests.
Evaluation, in addition to a right-sided cardiac catheterization, may include autoimmune, hepatitis, and HIV serology; ECG; echocardiogram with contrast to rule out shunting; 6-minute walk test; cardiopulmonary exercise stress test; polysomnography; ventilation/perfusion scan; chest CT angiography; and cardiac MRI3 (Table 1).
A vasodilator challenge in the cath lab is done to assess whether a patient is a candidate for calcium channel blockers since studies have shown increased survival in the long term for those who respond.4 The vasodilators typically used are inhaled nitric oxide, intravenous prostacyclin, or intravenous adenosine.
The respiratory therapist may be responsible for supplying nitric oxide to the cath lab and for the delivery system (face mask, oxygen tubing, etc). The physician may start the patient on as low as 10 ppm NO and increase after every 5 minutes until a maximum of 80 ppm has been met or a response is noted. A response is defined as having a decrease in mean PAP by >10 mm Hg to an absolute value of <40 mm Hg without a decrease in cardiac output.
Treatment
Treatment for PH, in general, focuses on the underlying cause; but PAH, in particular, tends to be idiopathic and is incurable. Consequently, drug therapy for PAH focuses on reducing vascular proliferation and augmenting vasodilation (Table 2). Prior to 1996, the median survival of patients with PAH was only 3 years.4

Table 2.
Combination therapy, utilizing multiple drugs to target all aspects of PAH, is the future of treatment strategies. Several studies have proven increased efficacy with a multidrug treatment plan.4
Once diagnosed and started on therapy, PAH patients are typically followed every 3 months utilizing the 6-minute walk test, with echocardiograms, BNP levels, and right heart catheterizations sometimes performed serially over time as well. It is recommended that PH patients be at least evaluated at some point by a specialized center that has experience with the complex diagnostic and treatment algorithms.
A 10% to 15% decline in the distance of the 6-minute walk test would indicate disease progression. The respiratory therapist needs to keep in mind other physical limitations and comorbid diseases the patient may have. For this reason, pulmonary rehabilitation is recommended.
Respiratory therapists are instrumental in the diagnosis and treatment of PH. Whether they are administering 6-minute walk tests or pulmonary function tests, providing nitric oxide or oxygen, helping to dispense iloprost or other respiratory therapy, or conducting pulmonary rehabilitation, respiratory therapists are likely to be involved with the care of these complicated patients.
Physicians rely on respiratory therapists to be competent in their testing and caregiving. Since PH is often diagnosed late, having skilled respiratory therapists involved will help ensure efficient diagnosis and treatment plans.
With special thanks to the Respiratory Therapy Department and Cardiac Catheterization Lab at Saint Joseph’s Hospital of Atlanta.
Stacy L. McLaughlin, BSRT, RRT, CPFT, is a pulmonary function specialist with Atlanta Pulmonary Group, Atlanta; Jeffrey E. Michaelson, MD, is a physician at Piedmont Pulmonary Consultants, Atlanta. For further information, contact [email protected].
References
- About Heart Failure. Available at: www.hearthope.com/about-heart-failure/nyha-scale.asp. Accessed August 20, 2010.
- Levine DJ. Diagnosis and management of pulmonary arterial hypertension: implications for respiratory care. Respir Care. 2006;51:368-81.
- McGoon MD, Kane GC. Pulmonary hypertension: disease and management. Mayo Clin Proc. 2009;84:191-207.
- Humbert M. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351:1425-36.